In our previous blogs, we discussed the changes already underway in the clinical evidence generation continuum and the associated rise in data mosaicism. The future state of evidence generation is also likely to undergo a significant shift as new technologies are incorporated into the medical product development lifecycle.
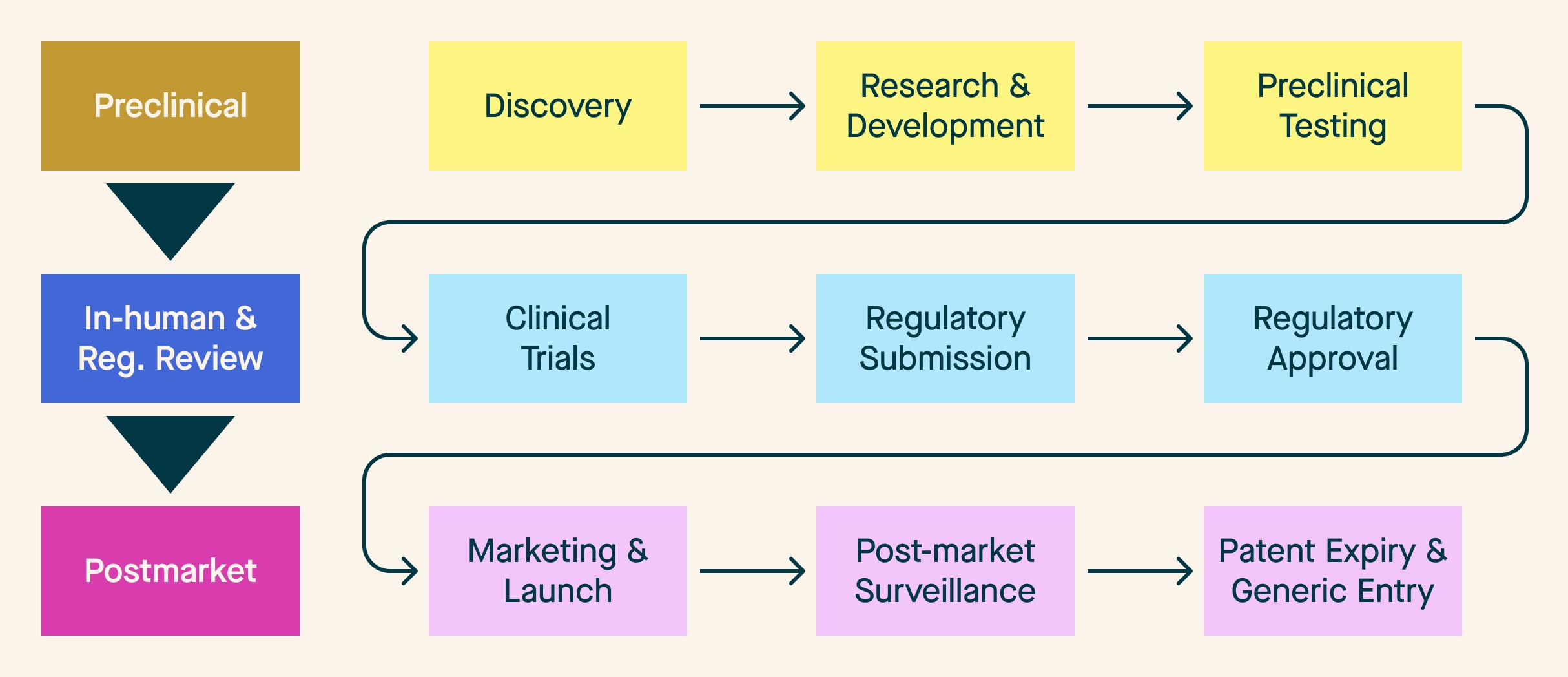
If we employ a traditional conception of a medical product lifecycle, we get something resembling Figure 1 - a multi-step, linear model with needs that fit into discrete, time-bound buckets.
Figure 1: Traditional Medical Product Lifecycle
As discussed in Part 1 of this blog series, companies are already taking advantage of new regulatory approaches and processes that shift some of the pre-market evaluation to the post-market setting. Even when taking advantage of these advances, the product lifecycle can be very lengthy and mean that 20 years can pass before a product is available to patients. We mapped this process using a typical timeframe for each “phase” in Figure 2.
Figure 2: Traditional Medical Product Lifecycle Timeline
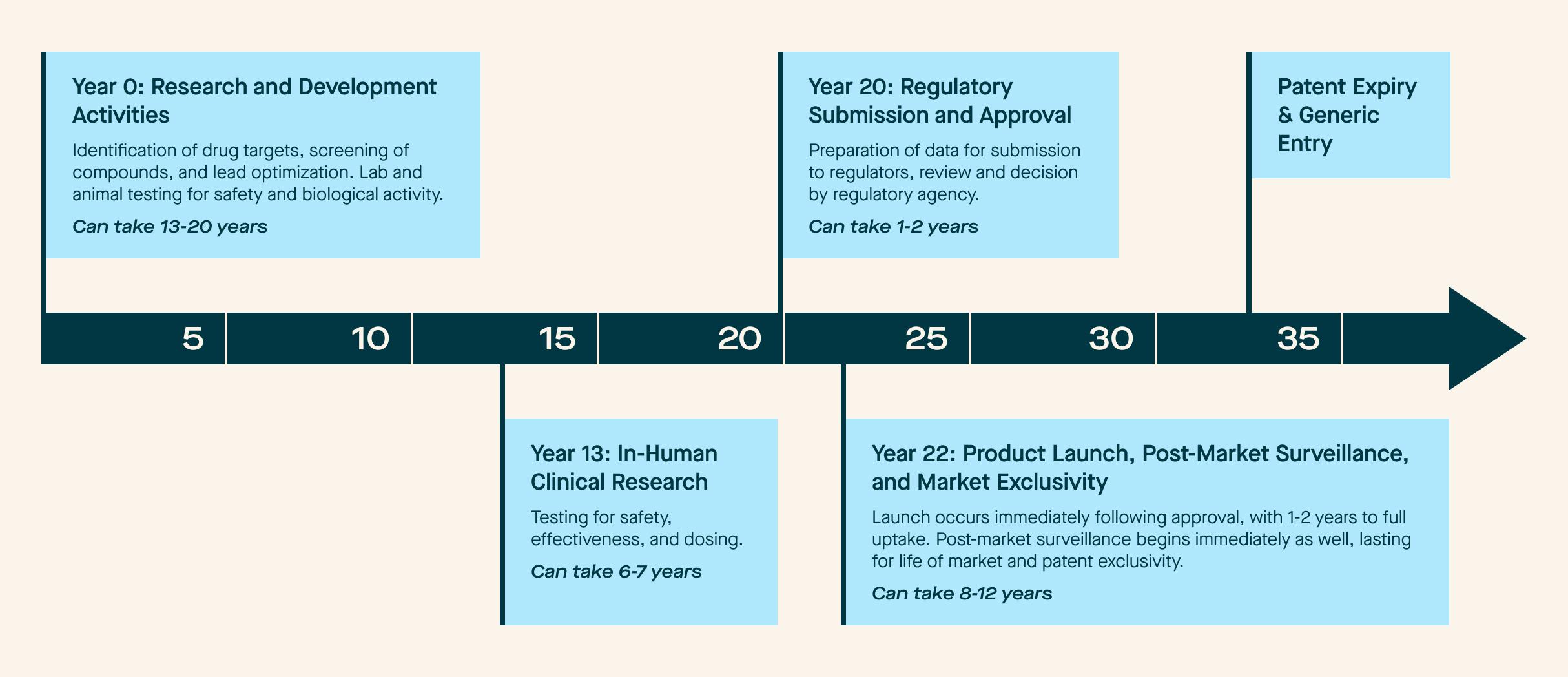
Not every product is the same, but this graph reflects the “traditional” state of play when it comes to research, development, approval, and maintenance. However, our repeated theme in this blog series is the imminent change coming to the industry. We are seeing technological and process evolutions that will dramatically modify this timeline in the coming years.
In the preclinical space, we are seeing rapid adoption of AI models to speed medical product discovery and target refinement, meaning drugs and biologics can enter more advanced testing faster than ever before. Accompanying this, we see regulators and researchers move away from animal testing in favor of organ-on-a-chip approaches and other new modalities such as in silico modeling and AI-enabled predictive toxicology. We’ve also seen companies seek out more favorable regulatory environments, moving preclinical and phase 1 activities overseas due to lower launch burdens.
Moving to the in-human clinical research and regulatory review phase, we observe increased adoption of adaptive trial designs and the incorporation of decentralized elements into clinical trials. These studies are stood up and completed faster, meaning data gets in front of regulators faster. Companies continue to take advantage of more favorable study environments in this research phase, as regulators are increasingly accepting data from overseas studies, as evidenced by FDA’s approval of four drugs in 2024 based on data from zero U.S.-based patients. While approaches such as synthetic control arms and real-world evidence generation may not have lived up to the original hype, we have seen companies use these approaches to enable approvals for products and indications that may not have made it to the market otherwise.
The postmarket phase is also seeing its fair share of innovation. As previously discussed, safety monitoring is increasingly shifting to the post-approval space and is aided by advancements in improved real-world data collection, abstraction, and curation. We are also seeing a rise in AI-enabled pharmacovigilance and digital health integration, resulting in faster signal detection and more nuanced data. Finally, we see payors embracing new modalities. For example, RWE is increasingly used to aid with formulary decision-making for biologic products and biosimilars.
It is not out of the question for these advancements in the aggregate to cut a product’s time to market in half (or even more!). We outline the potential future state in Figure 3.
Figure 3: Potential Future State of the Medical Product Lifecycle
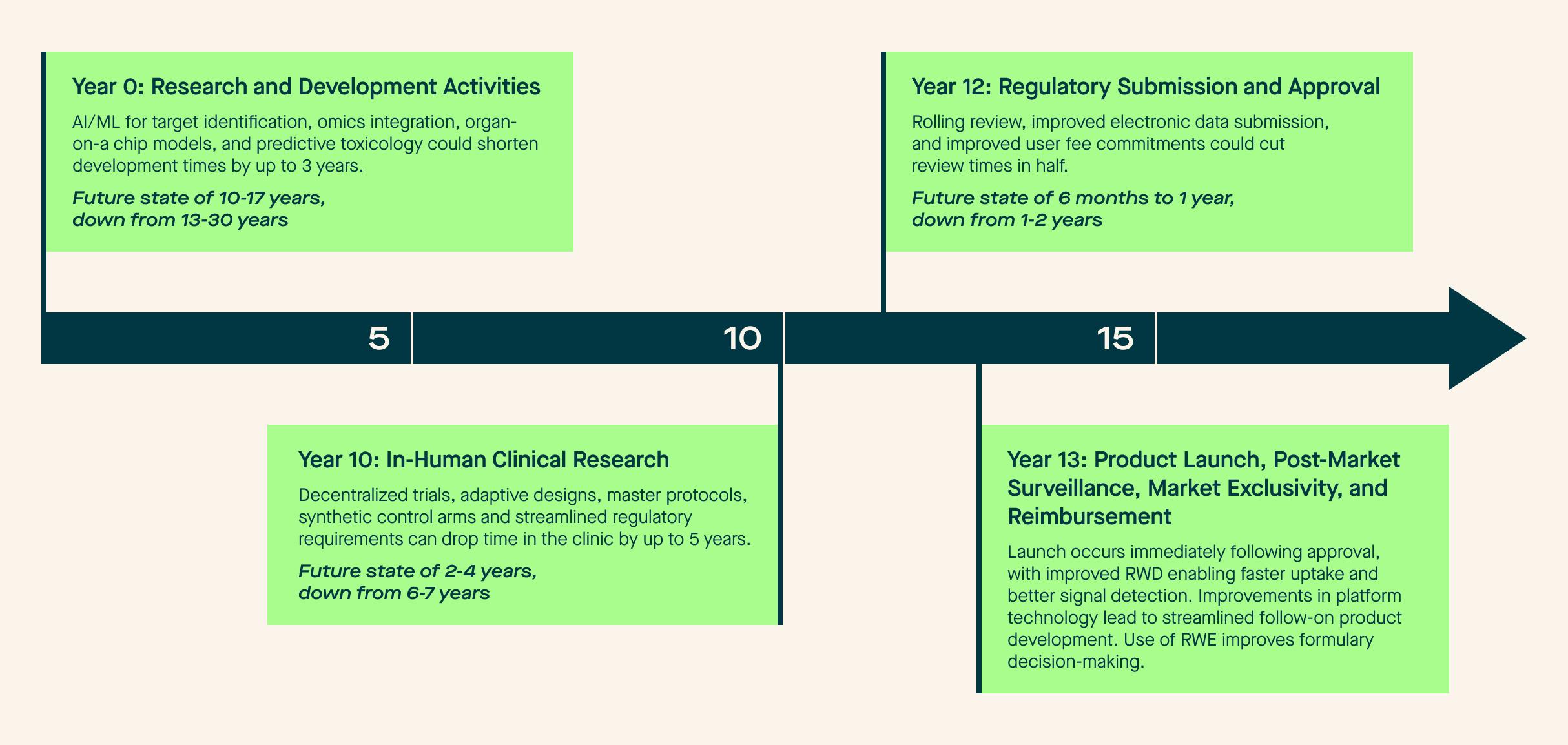
In order to achieve this future vision, we need to be good stewards of developing technology. Focusing specifically on AI-enabled data curation technologies, we recognize the need for solutions that solve for data quality when we incorporate the idea of mosaicism. When stitched together, these diverse sources can deliver a novel data infrastructure to advance clinical research. Longitudinal data from many different patients, aggregated into specific groups, can become cohorts. The industry and regulators must establish interoperability protocols that allow data to be effectively transferred between sources. Organizations will have to pay closer attention to metadata to understand how, when, where and from whom the information was collected.
Highlander Health understands that the real world is messy: not all clinically meaningful data fits neatly into rigid data schemas like USCDI. Improvements in large language models (“LLMs”) and NLP have created a window of opportunity to balance structure and flexibility in health data exchange. However, necessary guardrails must be in place. Any format must be non-proprietary to enable open access and prevent vendor lock-in. Additionally, all NLP or LLM transformations need to be auditable and transparent. This is easier said than done, as any LLM would have billions of parameters. This naturally points to the need for robust validation and quality control standards for AI tools in this arena. This idea of validation and quality control applies to all new modalities. Highlander Health is committed to developing learning labs that solve these problems and other major questions facing the evidence generation landscape. In our next blog in the “Mosaic Medicine” series, we will go deep on artificial intelligence and the crossroads we face in terms of validation and quality assurance.
Read Mosaic Medicine Part 3 on the Rise of Data Mosaicism here, Mosaic Medicine Part 2 on The Snail here, and Part 1 on A Vision of the Shifting Clinical Evidence Landscape here.
Mosaic Medicine
Institute | Resources
Public Policy
Technology